Note
Click here to download the full example code
Normalizing a cancer contact count matrix with CAIC¶
CAIC is a normalization method to remove copy-number biases present in a matrix. This example showcases how to perform such a normalization on simulated data with iced.
Loading the data and normalizing¶
The normalization is done in three step:
Normalize the data using LOIC, to remove GC, mappability, and other biases
Estimate the block biases due to copy number.
Remove the block biases from the LOIC-normalized contact counts
from iced import datasets
from iced import normalization
import matplotlib.pyplot as plt
from matplotlib import colors
counts, lengths, cnv = datasets.load_sample_cancer()
loic_normed = normalization.ICE_normalization(counts, counts_profile=cnv)
block_biases = normalization.estimate_block_biases(counts, lengths, cnv)
caic_normed = loic_normed / block_biases
Out:
/home/travis/build/hiclib/iced/iced/io/_io_pandas.py:56: UserWarning: Attempting to guess whether counts are 0 or 1 based
warnings.warn(
Estimating CNV-effects.
Computing relationship genomic distance & expected counts
Fitting Isotonic Regression
/home/travis/build/hiclib/iced/iced/normalization/_ca_utils.py:319: RuntimeWarning: invalid value encountered in double_scalars
bias[mask] = (c * c_exp).sum() / (c_exp**2).sum()
Computing relationship genomic distance & expected counts
Fitting Isotonic Regression
/home/travis/build/hiclib/iced/iced/normalization/_ca_utils.py:319: RuntimeWarning: invalid value encountered in double_scalars
bias[mask] = (c * c_exp).sum() / (c_exp**2).sum()
Computing relationship genomic distance & expected counts
Fitting Isotonic Regression
/home/travis/build/hiclib/iced/iced/normalization/_ca_utils.py:319: RuntimeWarning: invalid value encountered in double_scalars
bias[mask] = (c * c_exp).sum() / (c_exp**2).sum()
Computing relationship genomic distance & expected counts
Fitting Isotonic Regression
/home/travis/build/hiclib/iced/iced/normalization/_ca_utils.py:319: RuntimeWarning: invalid value encountered in double_scalars
bias[mask] = (c * c_exp).sum() / (c_exp**2).sum()
Computing relationship genomic distance & expected counts
Fitting Isotonic Regression
/home/travis/build/hiclib/iced/iced/normalization/_ca_utils.py:319: RuntimeWarning: invalid value encountered in double_scalars
bias[mask] = (c * c_exp).sum() / (c_exp**2).sum()
Computing relationship genomic distance & expected counts
Fitting Isotonic Regression
/home/travis/build/hiclib/iced/iced/normalization/_ca_utils.py:319: RuntimeWarning: invalid value encountered in double_scalars
bias[mask] = (c * c_exp).sum() / (c_exp**2).sum()
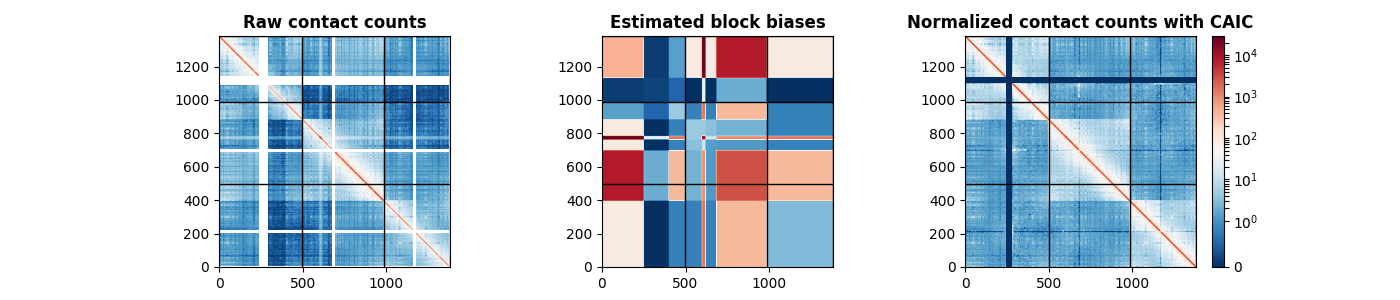
Visualizing the results using Matplotlib¶
The following code visualizes the raw original data, the estimated block biases, and the normalized matrix using the CAIC method.
chromosomes = ["I", "II", "III", "IV", "V", "VI"]
fig, axes = plt.subplots(ncols=3, figsize=(14, 3))
axes[0].imshow(counts, cmap="RdBu_r", norm=colors.SymLogNorm(1),
extent=(0, len(counts), 0, len(counts)))
[axes[0].axhline(i, linewidth=1, color="#000000") for i in lengths.cumsum()]
[axes[0].axvline(i, linewidth=1, color="#000000") for i in lengths.cumsum()]
axes[0].set_title("Raw contact counts", fontweight="bold")
m = axes[1].imshow(block_biases, cmap="RdBu_r", norm=colors.SymLogNorm(1),
extent=(0, len(counts), 0, len(counts)))
[axes[1].axhline(i, linewidth=1, color="#000000") for i in lengths.cumsum()]
[axes[1].axvline(i, linewidth=1, color="#000000") for i in lengths.cumsum()]
axes[1].set_title("Estimated block biases", fontweight="bold")
m = axes[2].imshow(caic_normed,
cmap="RdBu_r", norm=colors.SymLogNorm(1),
extent=(0, len(counts), 0, len(counts)))
[axes[2].axhline(i, linewidth=1, color="#000000") for i in lengths.cumsum()]
[axes[2].axvline(i, linewidth=1, color="#000000") for i in lengths.cumsum()]
cb = fig.colorbar(m)
axes[2].set_title("Normalized contact counts with CAIC", fontweight="bold")
Out:
Text(0.5, 1.0, 'Normalized contact counts with CAIC')
Total running time of the script: ( 0 minutes 21.815 seconds)